药明康德传媒报道
我们知道,有机分子大多由连接了氢原子的碳原子组成框架。如果能对碳氢键进行选择性的官能团化(functionalization),就能让人类充分利用“有机分子”这座金矿,在医药、农业、材料等领域带来巨大的应用价值。因此,科学界也称碳氢键活化为化学领域的圣杯。

▲本篇综述的通讯作者余金权教授(Credit: John D. & Catherine T. MacArthur Foundation.)
提到碳氢键活化,就不得不提美国加州Scripps研究所的著名华人学者余金权教授与他的团队。近年,这支团队在顶级学术期刊《科学》上连续发文,在碳氢键修饰的立体控制上取得了一系列进展。大年初一,大洋彼岸再次传来喜讯——今日,余金权教授团队在《科学》杂志上再度发文,在一篇深度综述中,系统地介绍了不对称碳氢键活化的立体化学模型的建立和设计可以加速碳氢键活化的手性过渡金属催化剂的理论基础。
传统手性有机合成主要集中于将二维平面碳原子中心转化为三维立体结构(例如不对称氢化,不对称环氧化等等)。基于C2对称模型的手性催化剂在工业界已经获得了广泛的应用,也因此被授予了2001年的诺贝尔化学奖。相比之下,基于从对称的三维四面体碳中心到手性的碳中心的官能团去对称化(desymmetrization)却由于底物的特殊性(需要基于官能团的对称面的存在)倍受局限。但是,如果我们将碳氢键视为可以反应的官能团,大多数有机分子中的多个碳原子往往存在基于碳氢键的对称面或镜面,因此通过碳氢键活化的去对称化将有广阔的应用前景。先前,余金权教授团队使用MPAA(mono-N-protected amino acid)配体,结合Pd(II)/Pd(0)催化循环,对含有吡啶导向基的底物进行去对称化,证明了其可行性。而MPAA配体容易获取和修饰的特性,更让人对其应用前景大为看好。
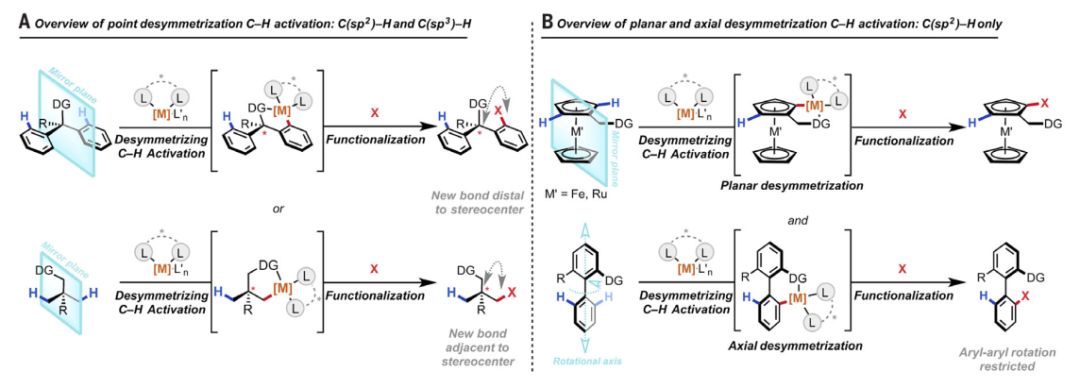
▲有机分子中的多个碳原子往往存在基于碳氢键的对称面或镜面(图片来源:《科学》)
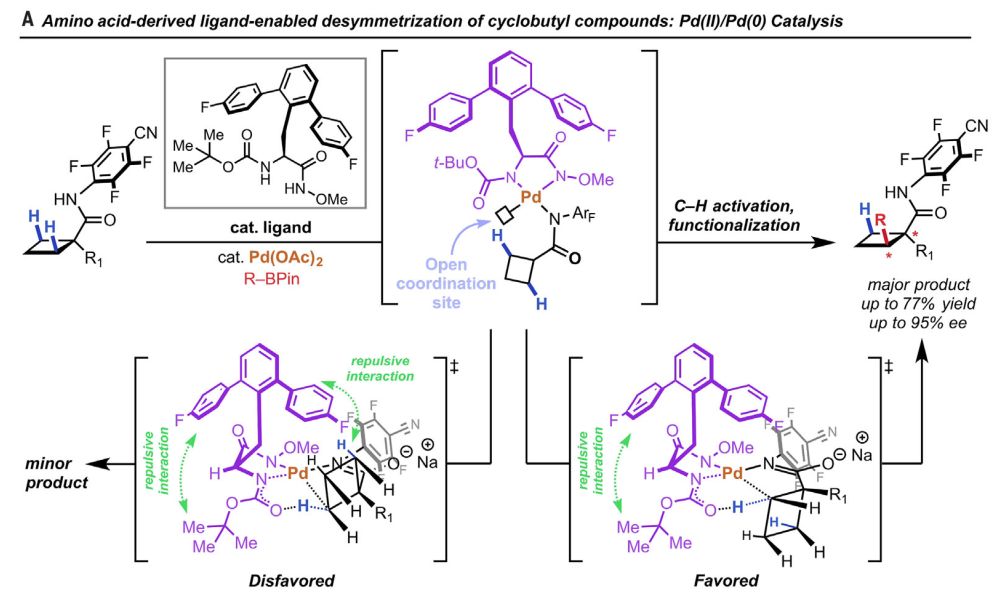
▲基于Pd(II)/Pd(0)催化,氨基酸衍生配体能对环丁基化合物进行去对称化(图片来源:《科学》)
虽然MPAA配体仍有很大的局限性,但是对其进一步的机理研究带来了新的思路。首先,经典的C2对称模型是设计手性催化剂的重要理论基础,但在碳氢键活化反应中,C2对称的双齿环丙基氨基酸配体却带来完全消旋的产物;其次,传统的导向碳氢键活化里,整个过程需要强配位导向基团(strongly coordinating directing groups)的参与。然而当导向基团过强时,不但会抑制后续的官能化过程,底物与过渡金属的结合能力还会超过配体与后者的结合能力,或使配体失效,或产生消旋的背景反应而得不到纯的手性分子。
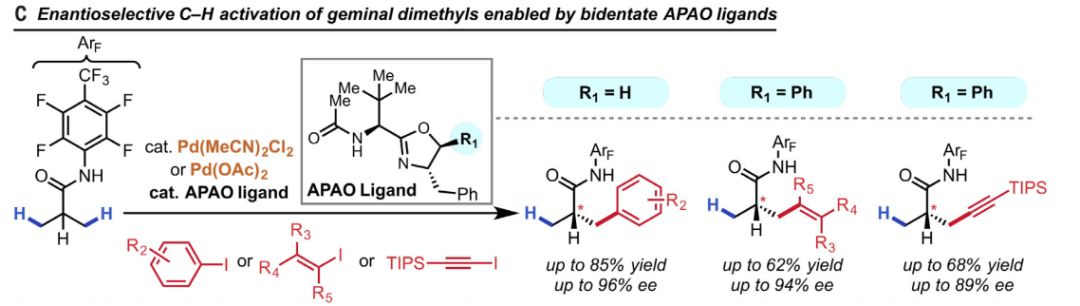
▲双齿APAO配体使偕二甲基发生立体选择性的碳氢键活化(图片来源:《科学》)
余金权教授团队借助这些原理提出了新的催化剂设计策略:非C2对称的双齿手性配体骨架和单齿弱配位的底物。依据这个理论框架,该团队合成了一种APAO(N-acetyl-protected aminomethyl chiral oxazoline)配体,能够对异丁酸衍生物进行立体选择性的碳氢键芳基化。进一步的探索则表明,这类配体同样能对异丁酸衍生物进行立体选择性的碳氢键烯基化和炔基化。值得欣喜的是,此类碳氢键的选择性官能团化有着良好的产率和很高的立体选择性。考虑到异丁酸的立体选择性羟化对于大量天然产物和药物分子的合成都异常重要,这一突破不仅能带来极好的应用前景,也能为碳氢键活化的设计与研究带来宝贵的经验。
在碳氢键活化中,亚甲基的立体选择性碳氢键活化一直被视为难点——为了在非环状底物里实现这一点,我们使用的催化剂必须能区分较小的氢原子与相对较大的R基团。而当R基团的大小逐渐接近氢原子时,对于催化剂的要求也会越来越高。
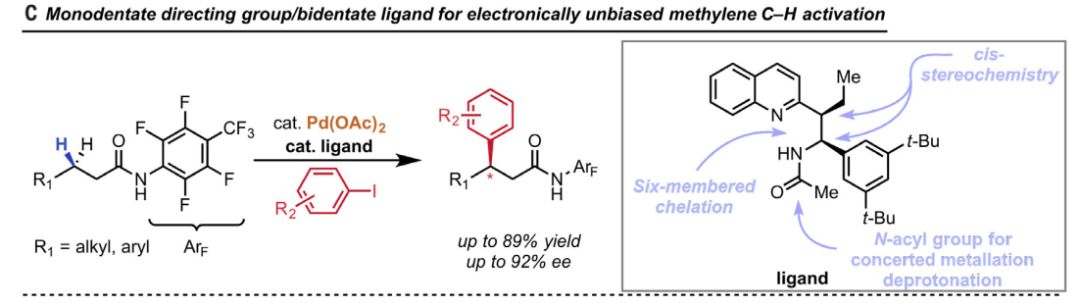
▲亚甲基立体选择性碳氢键活化的一个成功案例(图片来源:《科学》)
针对这一难点,余金权教授团队经过十四年如一日的坚守,终于开辟了一条全新的道路。先前,不少工作利用强双齿导向基团和单齿手性C2对称磷酰胺/磷酸,对苄基亚甲基进行碳氢键的活化。但和MPAA配体存在的问题一样,过强的导向基团会带来“背景反应”,这也影响到了此类反应的产率和立体选择性。为此,余金权教授的团队也已开始了全新配体的设计。先前的成功经验让他们意识到,喹宁配体能让Pd(II)催化的亚甲基碳氢键芳基化在弱导向基团下进行。系统性的配体修饰研究则表明,六元螯合是成功反应的关键,顺式取代则是高活性、高立体选择性的必须。基于这些发现和对非C2对称的要求,余金权教授团队找到了一款喹宁和氨基酸相接合的配体——它在C1位带有庞大的3,5-二叔丁基苯基,在C2位带有一个乙基。无论苄基还是普通烷基碳氢键,这款配体的独特设计均能促进亚甲基的碳氢键活化。这在配体开发和底物选择上,都给我们带来了诸多无价的洞见。
“2002年在剑桥开展研究金属不对称插入碳氢键时,主要是受好奇心驱使,并未意识到在药研方面的应用前景。有点小时候在山涧抓小鱼的感觉,实验手段非常原始但充满乐趣,”关于碳氢键活化的意义,余金权教授点评道:“现在看来,由于碳氢键的存在,有机分子的大多数碳中心都存在对称的魔镜让化学家们闯入一个充满对称性的分子世界。而打破这个魔镜所形成的手性碳金属键可以千变万化转化成所需要的官能团。考虑到手性是生命科学的基石之一,我猜想不对称碳氢键活化的应用将远远超出有机合成的范畴。”
在过去一个世纪里,不对称催化极大地改变了化学家们合成新分子的方式,也带来了大量药品、农用化学品、杀虫剂、新材料、以及其他自然产物。而理解不对称催化过程中的手性诱导,也是我们理解这些化学反应的重要工具。立体选择性的碳氢键活化正成为不对称催化的一片新天地,手性碳金属键多变的反应性能更是得天独厚,吸引了来自工业界和学术界的普遍兴趣。毫无疑问,这一大领域会突破极限,继续拓展,直到任何碳氢键都能以极高的产率和立体控制,自由转化为其他基团。这会是化学的一大里程碑。
我们再次祝贺余金权教授团队在碳氢键活化领域取得的一系列突破性成果,也期待未来能见证更多惊喜!
致谢:特别感谢余金权教授与本文作者之一朱如意的拨冗指正!
参考资料:
[1] St. Denis et al., Enantioselective C(sp3)–H bond activation by chiral transition metal catalysts, Science

